Sanne supports Casper met truien die ze vlak voor haar overlijden ontwierp
support u Sanne?
Sanne supports Casper met truien die ze vlak voor haar overlijden ontwierp
support u Sanne?






Een glossy tijdschrift over leven met alvleesklierkanker. Maar vooral ook over liefde, over troost en over verbinding. Doel is mensen informeren, ondersteunen en een hart onder de riem steken. Het circa honderd pagina’s tellende tijdschrift is gevuld met ervaringsverhalen, interviews met bijzondere personen als Dirk de Wachter en praktische zaken als voeding, beweging en mentale ondersteuning.
Het gedrukte magazine ‘Over leven met alvleesklierkanker’ is te bestellen door mensen die met de ziekte te maken krijgen. Een gedrukt exemplaar kan gratis worden opgevraagd door te mailen naar info@supportcasper.nl.
Wilt u de digitale versie bekijken? Laat hier uw mailadres achter en u ontvangt het magazine als PDF in uw inbox.
6 maart 2024
Leiden, 6 maart 2024 – Hoogleraar Chirurgie Casper van Eijck was ooit clubarts van Feyenoord, maar is vooral bekend om zijn strijd tegen alvleesklierkanker.
Dendritische Celtherapie
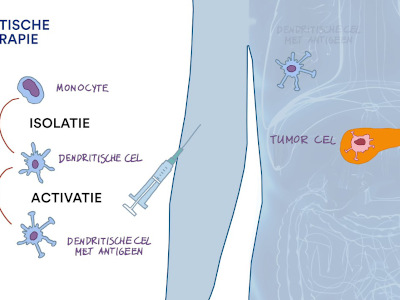
Animatie Dendritische celtherapie

Onderzoek Algemeen Support Casper 2023
![]() Support Casper is één van de goede doelen van Care-a-Lot, dé goede doelen loterij voor een gezonder Nederland. Als u meespeelt maakt u kans op mooie prijzen én steunt u maandelijks het onderzoek naar een behandelmethode van alvleesklierkanker, want minimaal 40% van uw inleg gaat naar ons.
Support Casper is één van de goede doelen van Care-a-Lot, dé goede doelen loterij voor een gezonder Nederland. Als u meespeelt maakt u kans op mooie prijzen én steunt u maandelijks het onderzoek naar een behandelmethode van alvleesklierkanker, want minimaal 40% van uw inleg gaat naar ons.
Start een actie
Support Casper evenementen
Word vrijwilliger
Word lid van de Business Club
Deel de campagne
Doneer éénmalig online
Doneer periodiek
Doneer via Tikkie
Periodieke schenking
Bedrijfssponsoring
Nalatenschap


Stichting OAK
Organisatie
Missie en visie
Verantwoording
Actueel
Timeline
Onderzoek
Gefinancierde studies
Pers
Contact
Stichting Overleven met Alvleesklierkanker
Bargelaan 200
2333 CW Leiden
071-7601620
info@supportcasper.nl
Contactformulier
KvK: 64583325
IBAN: NL82INGB0000042000
ANBI: 855730109
Cookie policy
Privacy verklaring
CBF erkenningsrapport
ANBI Verklaring
Klachten / Vertrouwenspersoon
Gedragscode
2015-2024 Support Casper Surlinio